Study of Density Functional Theory DFT of Amphiphilic Block copolymer poly(N-vinylpyrrolidone)-b-poly(4-vinyl benzene chloride)
N. Nemiche*, F. Z. Sebba
Department of chemistry, Laboratory of Macromolecular Chemical-Physical, Faculty of Science, University of Oran, Es Senia, El Menaouar B.P 1524 Oran31000.Algeria.
Abstract
The study of the synthesis of new chemical compounds are today often associated with a study by the methods of quantum mechanics, which use the distribution of electrons distributed in orbitals around the molecule, imply often high computing times which limit their use to small molecules or require the use of many approximations. The theoretical study "quantum chemistry" which is devoted to the modeling of the synthesis of the amphiphilic block copolymer poly (N-vinylpyrrolidone) -b-poly (4-vinyl benzene chloride) is based on DFT. This theoretical calculation aims to complete the description of this copolymer to be able to define the structure-property correlation. Thus, we use three different bases B3LYP, 6-31G and 6-31G ** to minimize energy and choose the best base. We will also discuss the results of the NMR, 13C NMR spectra and the IR spectrum.
Keywords: Amphiphilic, Block Copolymer, DFT, Quantum
Research and synthesis of new chemical compounds are now often associated with a molecular modeling study (Zhang et al., 2017; Liu et al., 2017). Molecular modeling is a technique that not only represents chemical properties and reactions but also manipulates two- and three-dimensional models of structures.
Molecular modeling involves the use of theoretical computation methods (molecular mechanics, molecular dynamics, ab-initio or semi-empirical quantum mechanics,...) to determine the graphical representation of the geometry or configuration of the atomic atoms, a molecule and evaluate the physicochemical properties of the molecule studied. Molecular modeling associated with an infographic representation of stereochemistry makes it possible to interpret physicochemical phenomena, to suggest new experiments and thus to analyze results in a more critical way than the experiments conventionally used, but these two purely theoretical approaches or experimental are complementary.
We propose to study with GAUSSIAN software (Bu and Zhang, 2016; Abu-Taleb et al., 2007; Nitta, 2009) Based on the fundamental laws of quantum mechanics, GAUSSIAN software allowed us to optimize energies, designated structural geometry and spectroscopic study. which relates to the vibrational frequencies of complex molecular systems the NMR study, and to anticipate their chemical properties.
To validate the method we base on the use of the gaussian software and its gauss view graphical side to model the molecule, the computation was launched with the correlation exchange functional B3LYP and the Gaussian base 6-31G + (p, d), where the set of functions ‘p’ added to the hydrogens and the set of functions ‘d’ added to the other heavy atoms is well defined.
At first, the theoretical calculation was made with two methods Hartree Fock (Jönsson et al., 2017; Khatri et al., 2016; Kanakaraju and Kolandaivel, 2002) and DFT (Wolny et al., 2016; Qin et al., 2014; Gómez-Pineda et al., 2010; Adrian-Scotto et al., 2010) using the functional B3LYP (Becke Lee-Yang-Parr).
Optimization with both methods gave us almost similar conformations, the last one having the lowest formation energy possible, hence the most stable conformation.
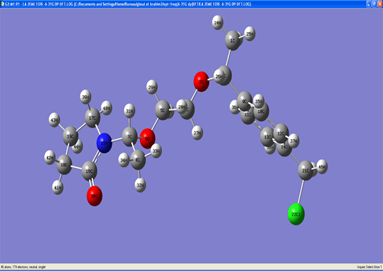
In our work, we have studied the theoretical part of the amphiphilic block copolymer already synthesize poly(N-vinylpyrrolidone) -b-poly (4-vinyl benzene chloride)
The scheme 1 represented the structure of the amphiphilic block copolymers.
Scheme 1. Structure of poly(4-vinyl benzene chloride)-b-poly-(N-vinyl pyrrolidone)
In this work, we performed a calculation of geometric and electronic parameters using the Gaussian software and its graphical face gauss view, and other quantum methods to model the molecule, the calculator was launched with the functional B3LYP correlation exchange and The Gaussian base 6-31G ** or 6-31G (p, d).
We have applied the most valid method "DFT ++", the different purely theoretical parameters of the Dipole Moment (DM), boundary orbital energy, HOMO, LUMO, and the energy gap are grouped in Table II.1.
Table 1: represents the different energy values of the dipole moment, the molecular orbitals HOMO, LUMO, and the energy gap.
|
dipole moment |
6.6955 Debye |
|
HOMO |
-0.03193 |
|
LUMO |
-0.24022 |
|
∆E |
0.20829 |
Boundary orbitals are two types of specific molecular orbital (OM) orbital: the highest occupied molecular orbital (HOMO) or HO (High Occupied) orbital which is the highest energy orbital occupied by at least one And the orbital LUMO (low unoccupied molecular orbital), BV (Low Vacante), which is the lowest orbital in energy not occupied by an electron. We carried out a qualitative study of the boundary orbitals of the copolymer geometry by the DFT method at the B3LYP / 6-31G level. The representation of the boundary orbitals HOMO and LUMO for the different isomers is shown in the following figures:
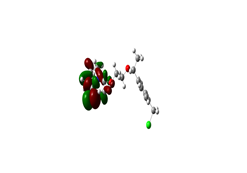
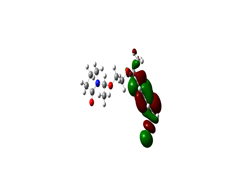
Figure 1: HOMO Image and LUMO 2D MESP (Map Electronic Surface Potential)
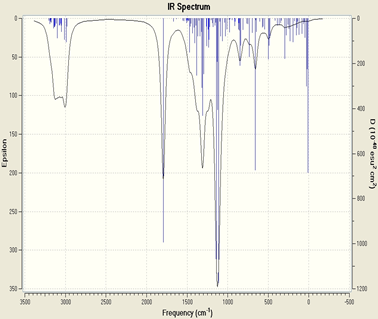
Examination of the infrared spectrum (Figure II.2) of the synthesized amphiphilic copolymer allowed us to identify the main absorption bands with:
A vibrating band located at 1138 cm -1 of the methoxyl group C-O, 1469 cm -1 which corresponds to the aromatic HC group, 2981 and 3010 cm -1 of the aliphatic CH group, 3133 cm -1 which corresponds to the NH group.
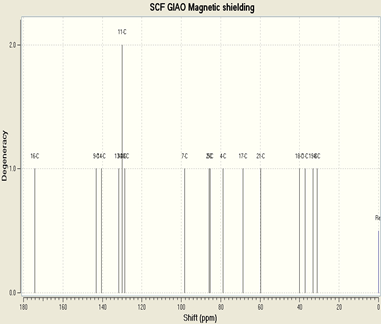
Regarding the 1H NMR, we observe a chemical shift which indicates methyl proton at 1 and 3ppm and other values slightly delineated of the methoxyl groups between 4 and 5 ppm, the hydrogens are aromatic proton about 7 and 8 ppm.
Figure 3:13C-NMR spectrum of poly (4-chloromethyl styrene) -b-poly (N-vinyl pyrrolidone) in CDCl3
the 13 C-NMR spectrum shows the chemical shifts of the methyl carbons to 31 and 33 ppm and the values 37 and 40 ppm indicate the carbons of the methoxyl group, while the aromatic carbons are represented by values between 98 and 120 ppm. The value 143 is C-N and 174 ppm carbonyl.
For the experimental part, it is noted that there is a lowering of value for the copolymer in the Infrared analysis from 2921 and 2881 cm -1 to 2974-3001 cm -1, also by 1H NMR analysis of 2.07 ppm to 1.75ppm,
This lowering of value in different spectra is due to the continuity of the chain in the copolymer concerning the monomer.
While a methyl at the end is more resonant of resonance or vibrational therms compared to ethyl in the middle of the chain which is of low vibration in IR as well as the chemical shift in NMR.
From the chemical environment, groups regardless of methoxyl or methyl as well as ethyls, there is a certain free rotation of the phenyl that it has the cone of shielding that at each random rotation these groups and which weakens the field B0 which crosses the nuclear population, which translates into a decrease in experimental chemical shifts.
Theoretical and experimental study of amphiphilic block copolymers thus constitutes a preliminary sketch for the development, characterization, and implementation of new polymeric materials that may have many potential applications in various fields (biomedical, pharmaceutical, textile, environmental). ...).
The theoretical study has made it possible to establish the structure-properties relationship of the copolymer, comparing experimental results already discussed and theoretical. Thus, the structure of the synthesized copolymer is optimized. The vibration modes are determined and compared with the experimental IR spectrum of the copolymer to validate our structure. To confirm the validity of the chosen model structure, the infrared spectrum of this model structure was simulated using the DFT / B3LYP-6-31G (d) method. It can be seen that the theoretical infrared spectrum of the optimized model structure correctly reproduces the experimental spectrum up to 1800 cm -1 with a fairly small shift between the vibration modes, which varies from 5 to 20 cm -1 depending on the bands. This shift can be explained mainly because the optimization of the structure is in the gaseous state, where the intermolecular interactions are absent whereas in the condensed state, case of the experiment, these interactions are present and strong. By combining experimental and theoretical studies, a structure-property correlation has been established.
This work was performed at the University of Oran in the laboratory of Chemical physical Macromolecular, and laboratory Modeling and Optimization of Industrial Systems, Faculty of Chemistry, University of USPTO, Oran, Algeria.
And thank’s of The co-authors wish to acknowledge Pr. F.Z.SEBBA and Pr .S.OULD KADA, for helping.
REFERENCES